Ajoene is a disulfide that has been found inA. sativumand has diverse biological activities, including antibacterial, anticancer, antiplatelet, and antioxidant properties.1,2,3,4It is active against Gram-positive (MICs = 5-160 µg ml) and Gram-negative bacteria (MICs = 136-200 µg ml), as well as yeasts (MICs = 10-20 µg ml).1Ajoene is cytotoxic to mouse melanoma cells (IC50= 18 µM), as well as human colon, lung, mammary, and pancreatic cancer cells (IC50s = 7-41 µM).2It reduces tumor growth in a B16 BL6 mouse model of melanoma when administered at a dose of 25 mg kg every other day and decreases the number of lung metastases when administered prior to tumor cell inoculation at doses ranging from 1-25 mg kg. It inhibits ADP- or collagen-induced platelet aggregation in isolated baboon platelets when used at concentrations ranging from 75 to 150 µg ml and in platelet-rich plasma isolated from baboons when administered at a dose of 25 mg kg.3Ajoene (25 mg kg) prevents thrombus formation on damaged arterial walls in heparinized pigs in anin situmodel of thrombogenesis.5It also reduces high-fat diet-induced hepatic steatosis, histopathological markers of liver damage, thiobarbituric acid reactive substances (TBARS) formation, and protein oxidation in a mouse model of non-alcoholic fatty liver disease (NAFLD).4 1.Naganawa, R., Iwata, N., Ishikawa, K., et al.Inhibition of microbial growth by ajoene, a sulfur-containing compound derived from garlicAppl. Environ. Microbiol.62(11)4238-4242(1996) 2.Taylor, P., Noriega, R., Farah, C., et al.Ajoene inhibits both primary tumor growth and metastasis of B16 BL6 melanoma cells in C57BL 6 miceCancer Lett.239(2)298-304(2006) 3.Teranishi, K., Apitz-Castro, R., Robson, S.C., et al.Inhibition of baboon platelet aggregation in vitro and in vivo by the garlic derivative, ajoeneXenotransplantation10(4)374-379(2003) 4.Han, C.Y., Ki, S.H., Kim, Y.W., et al.Ajoene, a stable garlic by-product, inhibits high fat diet-induced hepatic steatosis and oxidative injury through LKB1-dependent AMPK activationAntioxid. Redox Signal.14(2)187-202(2011) 5.Apitz-Castro, R., Badimon, J.J., and Badimon, L.A garlic derivative, ajoene, inhibits platelet deposition on severely damaged vessel wall in an in vivo porcine experimental modelThromb. Res.75(3)243-249(1994)
Oleic acid-13C is intended for use as an internal standard for the quantification of oleic acid by GC- or LC-MS. Oleic acid is a monounsaturated fatty acid and a major component of membrane phospholipids that has been found in human plasma, cell membranes, and adipose tissue.1,2 It contributes approximately 17% of the total fatty acids esterified to phosphatidylcholine, the major phospholipid class in porcine platelets.1 Oleic acid inhibits collagen-stimulated platelet aggregation by approximately 90% when used at a concentration of 10 μg ml. It also inhibits fMLF-induced neutrophil aggregation and degranulation by 55 and 68%, respectively, when used at a concentration of 5 μM, similar to arachidonic acid .3 Oleic acid (60 μM) induces release of intracellular calcium in human platelets.4
Psychotridine is an alkaloid that has been found inP. forsterianaand has diverse biological activities.1,2,3It inhibits ADP-, collagen-, or thrombin-induced aggregation of washed isolated human platelets with IC50values of 1.4, 1.4, and 3.9 μM, respectively.1Psychotridine (2.5 or 5 μM) is cytotoxic to HTC rat hepatocellular carcinoma cells.2It reduces paw licking induced by capsaicin in mice when administered at doses of 0.5, 2.5, or 5 mg kg.3 1.Beretz, A., Roth-Georger, A., Corre, G., et al.Polyindolinic alkaloids from Psychotria forsteriana. Potent inhibitors of the aggregation of human plateletsPlanta Med.51(4)300-303(1985) 2.Roth, A., Kuballa, B., Bounthanh, C., et al.Cytotoxic activity of polyindoline alkaloids of Psychotria forsteriana (Rubiaceae) (1)Planta Med.6450-453(1986) 3.Amador, T.A., Verotta, L., Nunes, D.S., et al.Involvement of NMDA receptors in the analgesic properties of psychotridinePhytomedicine8(3)202-206(2001)
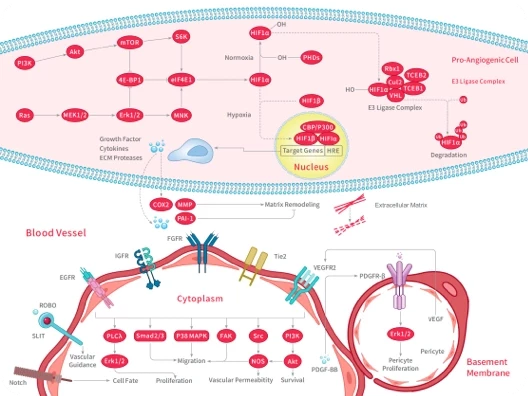
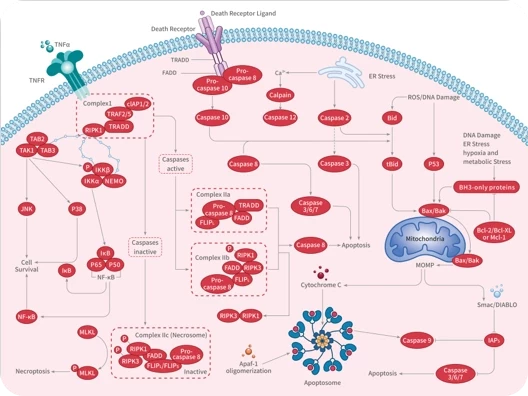
RWJ-56110 dihydrochloride is a potent, selective, peptide-mimetic inhibitor of PAR-1 activation and internalization (binding IC50=0.44 uM) and shows no effect on PAR-2, PAR-3, or PAR-4. RWJ-56110 dihydrochloride inhibits the aggregation of human platelets induced by both SFLLRN-NH2 (IC50=0.16 μM) and thrombin (IC50=0.34 μM), quite selective relative to U46619 . RWJ-56110 dihydrochloride blocks angiogenesis and blocks the formation of new vessels in vivo. RWJ-56110 dihydrochloride induces cell apoptosis[1][2]. Proteinase-activated receptors (PARs) are a family of G protein-coupled receptors activated by the proteolytic cleavage of their N-terminal extracellular domain, exposing a new amino terminal sequence that functions as a tethered ligand to activate the receptors.RWJ56110 inhibits the aggregation of human platelets induced by both SFLLRN-NH2 (IC50=0.16 μM) and thrombin (IC50=0.34 μM) while being quite selective relative to collagen and the thromboxane mimetic U46619 [1].RWJ-56110 dihydrochloride is fully inhibits thrombin-induced RASMC proliferation with an IC50 value of 3.5 μM. RWJ-56110 dihydrochloride shows blockade of thrombin's action with RASMC calcium mobilization (IC50=0.12 μM), as well as with HMVEC (IC50=0.13 μM) and HASMC calcium mobilization (IC50=0.17 μM)[1].RWJ56110 (0.1-10 μM; 24-96 hours) inhibits endothelial cell growth dose-dependently, with half-maximal inhibitory concentration of RWJ56110 is approximately 10 μM[2].RWJ56110 (0.1-10 μM; 6 hours) inhibits DNA synthesis of endothelial cells in a thymidine incorporation assays. Endothelial cells are in fast-growing state (50-60% confluence), RWJ56110 inhibits cell DNA synthesis in a dose-dependent manner, but when cells that are in the quiescent state (100% confluent), the inhibitory effect of PAR-1 antagonists is much less pronounced[2].RWJ56110 (0.1-10 μM; pretreatment for 15 min) inhibits thrombin-induced Erk1 2 activation in a concentration-dependent manner. However, when endothelial cells are stimulated by FBS (final concentration 4%), it reduces partially the activated levels of Erk1 2[2].RWJ56110 (30 μM; 24 hours) has an inhibitory effect on endothelial cell cycle progression. It reduces the percentage of cells in the S phase, while alterations in the percentages of G1 and G2 M cells are less pronounced[2]. Western Blot Analysis[2] Cell Line: Endothelial cells [1]. Andrade-Gordon, et al.Design, synthesis, and biological characterization of a peptide-mimetic antagonist for a tethered-ligand receptor. oc Natl Acad Sci U S A. 1999 Oct 26;96(22):12257-62. [2]. Panagiota Zania, et al. Blockade of angiogenesis by small molecule antagonists to protease-activated receptor-1: association with endothelial cell growth suppression and induction of apoptosis. J Pharmacol Exp Ther. 2006 Jul;318(1):246-54.
Isogarcinol is a natural polyisoprenylated benzophenone first isolated from plant species in the genus Garcinia. It has immunosuppressant actions, inhibiting the protein phosphatase calcineurin (IC50 = 36 μM) and suppressing the proliferation of T cells. Oral administration of isogarcinol in mice decreases delayed type hypersensitivity, prolongs graft survival in allogeneic skin transplants, suppresses inflammation in collagen-induced arthritis, and reduces clinical symptoms in experimental autoimmune encephalomyelitis. Isogarcinol inhibits the proliferation of HL-60 and PC-3 cancer cells (IC50s = 4 and 8 μg ml, respectively) through cell cycle arrest and apoptosis.
Pal-KTTKS is a lipidated pentapeptide consisting of a fragment of the type I collagen C-terminal propeptide conjugated to palmitic acid .1 It increases collagen production in human corneal and dermal fibroblasts when used at concentrations of 0.002, 0.004, and 0.008 wt%.2 Following topical administration, pal-KTTKS (50 μg/cm2) is found in the stratum corneum, epidermis, and dermis of isolated hairless mouse skin.1 It can self-assemble into flat tapes and extended fibrillar structures.3 Pal-KTTKS has been detected in anti-wrinkle creams.4 |1. Choi, Y.L., Park, E.J., Kim, E., et al. Dermal stability and in vitro skin permeation of collagen pentapeptides (KTTKS and palmitoyl-KTTKS). Biomol. Ther. (Seoul) 22(4), 321-327 (2014).|2. Jones, R.R., Castelletto, V., Connon, C.J., et al. Collagen stimulating effect of peptide amphiphile C16-KTTKS on human fibroblasts. Mol. Pharm. 10(3), 1063-1069 (2013).|3. Castelletto, V., Hamley, I.W., Whitehouse, C., et al. Self-assembly of palmitoyl lipopeptides used in skin care products. Langmuir 29(29), 9149-9155 (2013).|4. Chirita, R.-I., Chaimbbault, P., Archambault, J.-C., et al. Development of a LC-MS/MS method to monitor palmitoyl peptides content in anti-wrinkle cosmetics. Anal. Chim. Acta 641(1-2), 95-100 (2009).
Collinin is a coumarin that has been found in Z. schinifolium and has diverse biological activities.1,2,3,4 It is active against drug-susceptible and -resistant strains of M. tuberculosis (MIC50s = 3.13-6.25 μg/ml).1 Collinin inhibits LPS-induced nitric oxide (NO) production (IC50 = 5.9 μM) and reduces COX-2 protein levels in RAW 264.7 cells.2 It completely inhibits aggregation of isolated rabbit platelets induced by arachidonic acid , collagen, or platelet activating factor (PAF) when used at a concentration of 100 μM.3 Dietary administration of collinin (0.05% w/w) reduces the number of mice with tumors and the number of tumors per mouse in a mouse model of colitis-related carcinogenesis.4 |1. Kim, S., Seo, H., Al Mahmud, H., et al. In vitro activity of collinin isolated from the leaves of Zanthoxylum schinifolium against multidrug- and extensively drug-resistant Mycobacterium tuberculosis. Phytomedicine 46, 104-110 (2018).|2. Nguyen, P.-H., Zhao, B.T., Kim, O., et al. Anti-inflammatory terpenylated coumarins from the leaves of Zanthoxylum schinifolium with α-glucosidase inhibitory activity. J. Nat. Med. 70(2), 276-281 (2016).|3. I.S., C., Lin, Y.C., Tsai, I.L., et al. Coumarins and anti-platelet aggregation constituents from Zanthoxylum schinifolium. Phytochemistry 39(5), 1091-1097 (1995).|4. Kohno, H., Suzuki, R., Curini, M., et al. Dietary administration with prenyloxycoumarins, auraptene and collinin, inhibits colitis-related colon carcinogenesis in mice. Int. J. Cancer 118(12), 2936-2942 (2006).
MMP13-IN-2 is a highly potent, selective, and orally active inhibitor of MMP-13. It demonstrates exceptional potency against MMP-13, with an IC50 value of 0.036 nM, and exhibits selectivities greater than 1,500-fold over MMP-1, 3, 7, 8, 9, 14, and TACE. Moreover, MMP13-IN-2 possesses the capability to effectively inhibit collagen release from cartilage in vitro. Consequently, MMP13-IN-2 holds great potential for advancing research on collagenase-related diseases.
Collagen-IN-1 (compound 3) is an ortho-carbonyl hydroquinone derivative exhibiting selective inhibition of collagen. It non-competitively inhibits agonist-induced platelet aggregation, with an IC50 value of 1.77 μM. Collagen-IN-1 effectively reduces the expression of P-selectin, activation of glycoprotein IIb IIIa, and release of adenosine triphosphate and CD63 from platelets. This compound holds promise for research into platelet-related thrombosis diseases [1].
GPVI antagonist 3 (Compound 2) is a potential antagonist that shows promising antiplatelet activity by selectively inhibiting the interaction between the Glycoprotein VI (GPVI) receptor and its ligands. It demonstrates potent inhibitory effects, with IC50 values of 1.01 μM for collagen, 1.92 μM for CRP, 7.24 μM for convulxin, and 51.74 μM for thrombin. GPVI is a major collagen receptor on platelets, making it an ideal target for safe and effective antithrombotic treatment. Therefore, GPVI antagonist 3 holds potential as a novel antiplatelet agent [1].
Angiotensin II human (Angiotensin II) TFA 作为肾素 血管紧张素系统中关键的生物活性肽,扮演着血管收缩剂的角色并在调节人体血压中发挥中心作用。其主要通过与 G 蛋白偶联受体 (GPCRs)、血管紧张素 II 1型受体 (AT1R) 和血管紧张素 II 2型受体 (AT2R) 的相互作用来介导效应,包括刺激交感神经系统、增加醛固酮的生物合成和肾脏功能。此外,Angiotensin II human TFA 促进血管平滑肌细胞的生长和 I 型及 III 型胶原在成纤维细胞中的合成,导致血管壁与心肌增厚及纤维化,并诱导细胞凋亡。还通过LOX-1依赖的氧化还原敏感路径诱导内皮细胞中的毛细血管形成。
(±)17(18)-EpETE-Ethanolamide, an ω-3 endocannabinoid epoxide, originates from eicosapentaenoic ethanolamide (EPEA) through cytochrome P450 (CYP) epoxygenases action and is decomposed by soluble epoxide hydrolase (sEH) and fatty acid amide hydrolase (FA, AH). Its endogenous synthesis occurs in LPS-stimulated and EPEA-supplemented BV-2 microglia cells, a process inhibited by the CYP inhibitor ketoconazole. This compound mitigates IL-6 and nitrite levels while enhancing IL-10 production following LPS exposure in BV-2 microglia. At a dose of 50 µM, it prevents platelet aggregation caused by arachidonic acid but not that triggered by ADP, collagen, or ristocetin. Additionally, it facilitates the dilation of constricted bovine coronary arteries (ED50= 1.1 µM) and blocks VEGF-driven tubulogenesis in human microvascular endothelial cells (HMVECs).




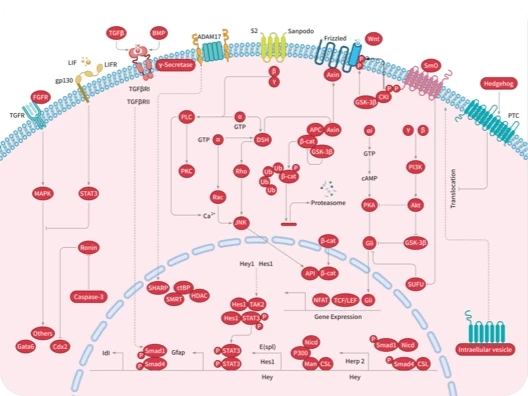
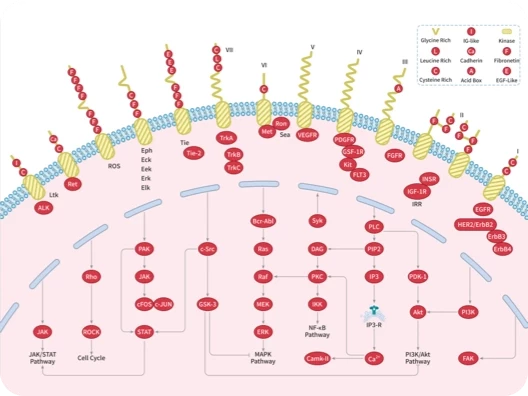
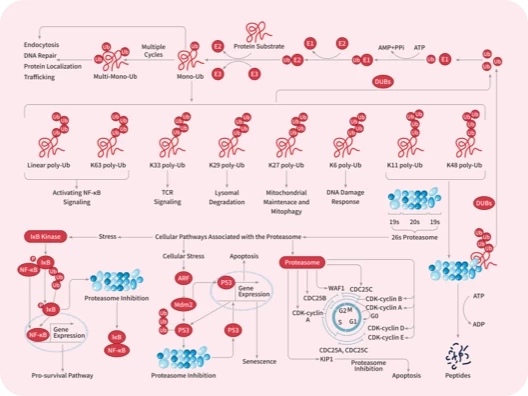
 您的购物车当前为空
您的购物车当前为空












 嗨!有任何问题?点我咨询
嗨!有任何问题?点我咨询